HPLC分析方法开发与验证系统全面讲解
一、分析方法开发
分析方法的开发主要包括色谱柱的选择、流动相的选择、检测波长的选择和梯度的优化几个方面。目前高效液相多做反相使用,所以本文主要以反相为例进行讲解。
1.色谱柱的选择
原料药生产对产品的纯度和杂质含量的要求非常苛刻,要求检测使用的色谱柱有较高的理论塔板数,能提供更好的分离度,从而对可能存在的杂质有更大的分离的可能性,所以5um填料的色谱柱长要250mm,3.5um填料的柱长要150mm,基本上都是各个粒径柱长最长的。我比较喜欢近两年新出的亚二微米填料的色谱柱,50mm柱长就能提供很高的理论塔板数,而且柱长和粒径小了,流速增加很多,能节省很多的分析时间,极大的提高工作效率。一般选用直径为4.6mm或3.0mm的柱子,太细了可能会增大柱外效应。填料的孔径对于小分子合成药物不需要考虑,普通的分析柱都在100A左右,能满足分析检测的需要。
对于API分析方法开发,一般要求必须做色谱柱的筛选实验,最少使用三种不同类型的色谱柱,每种类型三只,要来自于不同厂家。
三种类型包括:
1)普通的C18或相应的C8色谱柱,如Waters的Symmetry C18或C8,YMC的Pack Pro C18或C8,Agilent的RX C8等,其它公司如菲罗门和热电也有相应的色谱柱;
2)封端处理的或者极性嵌入型色谱柱,如Waters的SymmetryShield RP18或RP8,XTerra RP18或RP8,YMC的ODS AQ,Agilent的Zorbax SB AQ等,其它公司如菲罗门和热电也有相应的色谱柱;
3)填料用其它官能团修饰过的色谱柱,如苯基柱等,很多公司都有。
一般不同类型的色谱柱在选择性上会有很大的差异,相同类型的色谱柱生产厂家不同在选择性上也会有差异,这个主要是填料的性质和生产工艺决定的,有时候用一只色谱柱分离不好,除了优化梯度和流动相外,换一个厂家的柱子也是一个很好的选择。相同品牌型号的色谱柱,C18和C8在选择性上没有差异,但是C18保留能力更强,相同的样品分离度更高,我们一般倾向于选择用C18。我们在筛选色谱柱时尽量选择行业内排名前几位的厂家,柱子品质好,开发分析方法时能省很多力气,做出来的分析方法也有保证。一个药从开发到上市可能会持续十几年甚至更长时间,厂家有实力,开发方法时选定的柱子在若干年以后需要时还会有的买,做分析时重复性也能保障。多用几只色谱柱做筛选和分析方法优化,能尽最大的可能提高分析方法的质量,保证检测结果的可信度。
我比较喜欢用的柱子有:Agilent的Zorbax EclipseXDB-C18、ZorbaxEclipse Plus C18,Waters的Symmetry C18、XTerra RP18、XTerra MS C18等,YMC的柱子有时会是不错的备用选择,菲罗门的柱子菲罗门的柱子国内外市场占有率较高,但是感觉柱子压力高,寿命短,尽量不用,热电的柱子用的也不少,但没什么感觉。
制剂分析方法选择色谱柱的要求基本和API相一样,对于中间体和生产过程中反应跟踪(IPC)分析方法开发使用的色谱柱,我们一般会根据样品性质直接选取一两只普通C18或者封端处理过的色谱柱,简化筛选过程。
2.流动相的选择
常用做反相流动相的溶剂是甲醇和乙腈,甲醇有其性价比的优势,但是甲醇活性高,可能与某些样品发生反应,而且甲醇在低波长下有紫外吸收,会降低分析方法的灵敏度;乙腈虽然价格很高,毒性比甲醇大,但是洗脱能力比甲醇强,很少与样品发生反应,用作流动相系统压力要比甲醇低很多,且截止波长比甲醇低20nm,增加了检测出在低波长下才有吸收的杂质的可能性,所以我们一般倾向于多用乙腈,少用甲醇。但是有时候样品峰形不好或者分离不好,更换溶剂试试是一个很好的选择,毕竟不同的溶剂提供不同的选择性。
对流动相的优化主要在水相上下功夫,水里可以加酸、加碱、加盐,从而改善峰形、提高分离度。流动相里加碱的情况比较少,主要还是加酸,常用的酸有磷酸、三氟乙酸、甲酸、乙酸、高氯酸、甲基磺酸等,其中最常用的是磷酸和三氟乙酸,磷酸在低波长下没有紫外吸收,而三氟乙酸在低波长下有,但是三氟乙酸易挥发而磷酸不行,所以单纯做液相,低波长下磷酸最合适,三氟乙酸有吸收,运行梯度时基线漂移很严重,而做液质就要考虑首选三氟乙酸了,近些年还比较流行加甲酸或乙酸。一般情况下这几种酸没有太大区别,我们更多的是考虑通过加酸改变流动相的pH值,从而改善样品的分离度和峰形。下面两图是流动相的pH值改变对一组样品分离和峰形的影响。
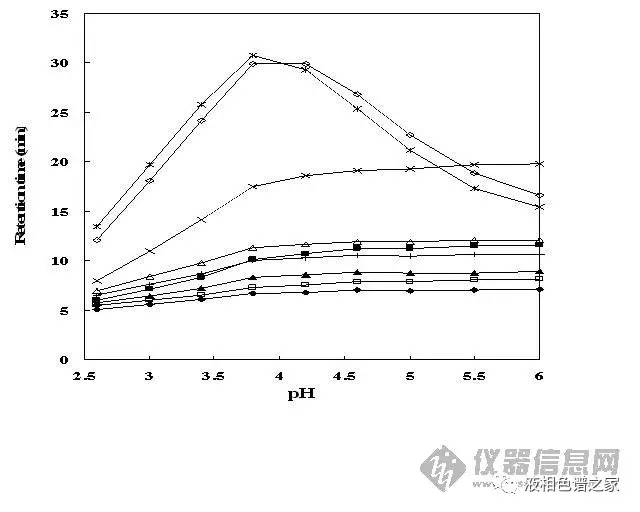
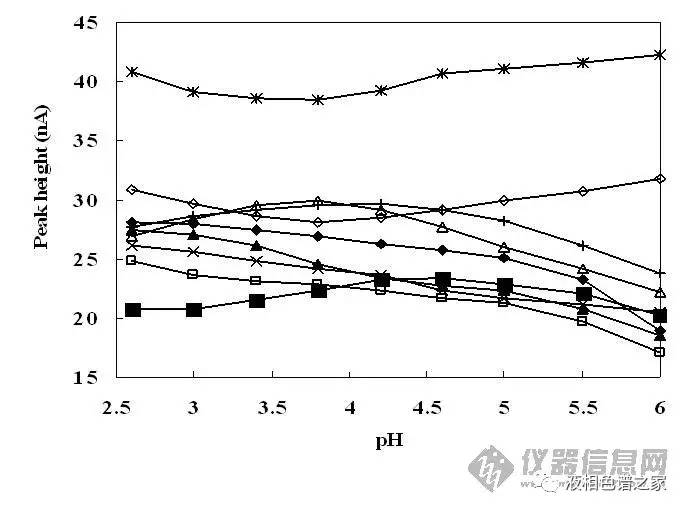
从图中可以看出:
1)流动相pH值改变可以改变样品的保留时间和分离度;
2)流动相pH值改变甚至可以改变某些样品出峰的先后顺序;
3)流动相pH值改变可以改变样品的峰高,即可以调整峰形。
相同进样量样品峰越高则意味着峰形越好,从图中可以看出多数样品在低pH值下峰形都比中性要好,这个主要是由色谱柱本身的性质所决定的。色谱柱主要都是硅胶基质,现有的填料处理工艺无法将硅胶上残余的硅羟基全部去除,硅羟基会造成样品峰拖尾,一般认为硅羟基的pKa在3.5到4.5之间,低pH值能帮助抑制硅羟基的活性,减小拖尾,从而改善峰形,提高分离度。水溶液中添加0.1%(体积)的磷酸或者三氟乙酸其pH值大概在2左右,用作流动相正好抑制硅羟基的活性,所以开发液相分析方法时流动相首选水加01.%的磷酸,然后再以此为基础做优化。
在单独用酸不行的时候就要考虑使用缓冲盐,缓冲盐的选择原则是:简单、稳定、缓冲能力强、配制简单,需要调pH值时要有相应的酸或碱。常用的缓冲盐是磷酸盐,主要是钾盐和钠盐,再有就是醋酸盐,常用的盐浓度在10~20mM左右。以前因为色谱柱填料生产工艺的问题,往往需要在流动相里添加三乙胺来减少拖尾,但是三乙胺对色谱柱的寿命有很大影响,现在新的色谱柱都不再需要了。流动相里有时会需要调节pH值到碱性,具体pH要视色谱柱的耐受范围而定,常用NaOH、KOH溶液或氨水做为调节缓冲盐溶液碱性pH的试剂,也可以往水里单独添加氨水做碱性流动相。
在缓冲盐做流动相时,出峰太早、峰形很差、相似结构的化合物峰因为拖尾或峰型太宽而不能达到基线分离时,可以考虑使用离子对试剂,常用的离子对试剂主要是各种烷基磺酸钠和四丁基铵盐,但是流动相里使用离子对试剂时,系统需要的平衡时间长,样品保留时间不是很稳定,因为离子对试剂的背景吸收基线会很差,且做完样品后需要长时间清洗,所以我们尽量不使用离子对试剂。
使用缓冲盐时要注意流动相混合以后盐可能析出的问题和盐背景吸收导致基线漂移严重的问题,尤其是在流动相里添加醋酸铵以后,在低波长下梯度变化时基线下降非常严重,严重影响对含量较小杂质的准确定量,可以考虑在乙腈里加入10%的水,水中预先加入10倍水溶液浓度的缓冲盐,这样梯度中A、B两项盐的浓度相同,可以避免基线漂移严重的问题。
原料药一般结构式比较大,分子构成比较复杂,开发分析方法时用水加磷酸效果可能效果不好,通常还要求最少尝试2和6.5两个pH值的磷酸盐缓冲溶液,并依据结果对流动相进行pH优化,如效果不理想再进一步尝试其它缓冲盐溶液。开发中间体或者IPC的分析方法时可以根据经验酌情简化流动相的选择过程。
3.梯度的优化
梯度优化主要是通过调节流动相的起始比例和梯度的斜率来调整样品的保留时间,优化样品的分离度。下图是梯度中有机相起始比例的变化对一组样品分离的影响。
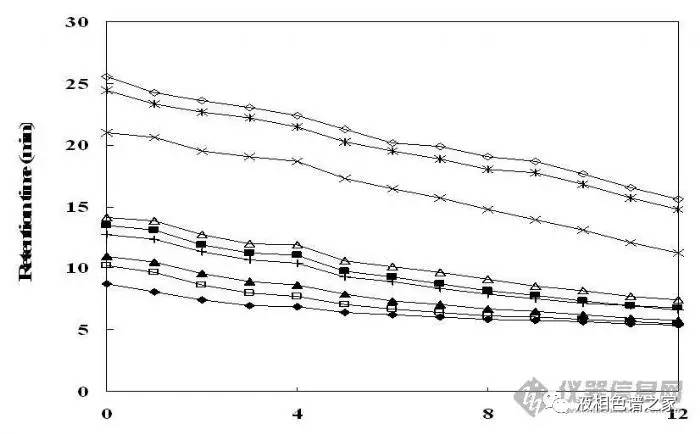
从图中可以看出:有机相起始比例越小,样品保留时间越长,随着梯度的改变,样品出峰的先后顺序也有可能改变。我们做梯度优化时主要调整梯度的起始比例和斜率。现在的色谱柱或者采用了新的封端工艺,或者内嵌极性基团,耐水的能力都比较高。在酸性条件下很多化合物都以离子形式存在,极性较大,为了提高样品的分离度,尽量使用大比例的水做梯度的起始。对于添加缓冲盐的流动相要注意梯度变化过程中流动相组成改变时不能有盐析出。
对于水加0.1%磷酸的流动相,开始时可以采用95%的水做起始,以95%的有机相结束,注意根据实际情况在梯度最后用大比例的有机相冲洗几分钟,以保证把小极性的杂质洗脱下来,防止样品残留到下一针。梯度的斜率一般采用凹线型的先小后大,梯度变化先慢后快,在此基础上再对梯度进行优化。
使用缓冲盐溶液的梯度水相起始比例一般要从10~20%开始,为了防止盐析出,在梯度最后避免用纯的有机相做冲洗,梯度斜率采用恒定的就可以,在此基础上根据方法运行的情况对梯度进行调整。
一般一个API的样品采集的时间控制在40~50分钟左右,样品出峰在15~20分钟左右比较好,如果有极性非常小的杂质存在可以在最后加一段时间的大比例有机溶剂冲洗色谱柱,最后再设置10分钟左右的重新平衡时间。中间体和IPC的样品分析方法时间可以根据需要减半或者时间更短。
4.波长的选择
做分析方法开发需要二极管阵列检测器,做色谱峰纯度检查和选择检测波长,通过色谱峰纯度检查来保证主峰里没有掩盖其它杂质,做纯度检测对波长选择的要求比较简单,原则是把尽量多的杂质在色谱图上体现出来。很多杂质只有在低波长下才有紫外吸收,所以我们选择尽可能低的波长,乙腈的截止波长在190~195nm,用乙腈做流动相检测波长可以选择在210~220nm。也有公司要求分别选取产品紫外吸收最强的波段和210~220nm两个波段做对比,哪个纯度低以哪个为准,这样可以更严格的控制产品的质量。
二、分析方法验证
为了保证分析检测结果准确、可靠,必须对所采用的分析方法的准确性、科学性和可行性进行验证,以证明分析方法符合检测的目的和要求,这就是分析方法验证。从本质上讲,方法验证就是根据检测项目的要求,预先设置一定的验证内容,并通过设计合理的试验来验证所采用的分析方法符合检测项目的要求。方法验证在质量控制上有重要的作用和意义,只有经过验证的分析方法才能用于药品生产的分析检测,方法验证是制订质量标准的基础。方法验证内容包括方法的专属性、线性、范围、准确度、精密度、检出限、定量限、耐用性和系统适用性等,检测目的不同验证要求也不尽相同。
1.专属性
专属性是指分析方法能够将产品和杂质分开的特性,也称为选择性。对于纯度检测,可在标准品中加入产品中的已知杂质,或者直接用粗品,考察产品峰是否受到杂质的干扰,对于过程跟踪,可用反应体系样品来考察有没有其它的杂质干扰。必要时使用二极管阵列检测器或者质谱检测器进行色谱峰纯度检查。一般要求产品和杂质之间的分离度大于2.0。
2.线性
线性是在设定的范围内,检测结果与样品中原料或产品的浓度呈线性关系的程度。线性是定量检测的基础,需要定量检测的项目都需要验证线性。一般用贮备液经过精密稀释,或分别精密称样,制备得到一系列被测物质的浓度(5个以上),按浓度从小到大运行序列,以峰面积和浓度的函数作图,用最小二乘法进行线性回归计算,考察分析方法的线性。
3.范围
范围指在能够达到一定的准确度、精密度和线性时,样品中被分析物的浓度区间。简单的说,范围就是分析方法适用的样品中待测物的浓度最大值和最小值。需要定量检测的分析方法都需要对范围进行验证,纯度检测时,范围应为测试浓度的80%~120%。
4.准确度
准确度是指测定的结果与真实值之间接近的程度,所以也叫做真实度,需要定量得分析方法均需要验证准确度。准确度应在规定的范围内建立,对于原料药可用已知纯度的标准品或符合要求的原料药进行测定,必要时可与另一个已建立准确度的方法比较结果。
5.精密度
精密度是指在规定条件下,同一均匀样品经多次取样进行一系列检测所得结果之间的接近程度。精密度一般用相对标准偏差表示,取样检测次数应至少6次。
精密度可以从三个层次考察:重复性、中间精密度、重现性。
a、重复性是在相同的操作条件下、较短时间间隔内,由同一分析人员测定所得结果的精密度。一般是用100%浓度水平的样品测定6次的结果进行评价。
b、中间精密度:同一实验室,在日期、分析人员、仪器等内部条件改变时,测定结果的精密度。
c、重现性:指不同实验室之间不同分析人员测定结果的精密度。
6.检出限
检出限是指样品中的被分析物能够被检测到的最低量,不需要准确定量。检出限体现了分析方法的灵敏度。检出限的测定可以通过对一系列已知浓度被测物的试样进行检测,以能准确、可靠检出被测物的最小浓度来确定,也可把已知浓度样品的信号与噪声信号进行比较,以信噪比为3:1时的浓度确定检出限,一般要求能够达到进样浓度的0.05%。
7.定量限
定量限是指样品中的被分析物能够被定量检测的最低量,其测定结果需要一定的准确度和精密度,定量限体现了分析方法灵敏定量检测的能力。检测需要严格控制含量的杂质,必须考察方法的定量限,以保证杂质能够被准确定量。一般以信噪比为10:1时相应的浓度或进样量来确定定量限。
8.耐用性
耐用性是指测定条件发生小的变动时,测定结果不受影响的承受程度,耐用性主要表明方法的抗干扰能力,主要的变动因素包括:流动相的组成、流速和pH值、色谱柱、柱温等。经试验,应说明小的变动能否符合系统适用性试验要求,以确保方法有效。
9.系统适用性试验
液相色谱分析方法主要依赖高效液相色谱仪和色谱柱,在做方法验证时,有必要将高效液相色谱仪、色谱柱、流动相与实验操作、待测样品等一起当作完整的系统进行评估,并将系统适用性作为分析方法的组成部分,系统适用性便是对整个系统进行评估的指标。一般系统适应性的要求为:分析方法能够达到0.05%的检出限,主峰的拖尾因子0.5<Tf<2.5,主峰与杂质的分离度大于2.0,空白干净,主峰处无系统峰干扰。
三、总结
分析方法开发与验证是一个整体,在实际工作中,一般是先开发分析方法,经过适当的优化以后再做方法验证,验证的部分内容在分析方法开发时就要做,比如说分析方法的专属性验证。分析方法验证并非必须验证所有的内容,只要注意验证内容充分,足以证明分析方法的合理性就可以了,如杂质限度检测一般只需要验证专属性和检出限,而精密度、线性、定量限等涉及定量测定的项目,则不需要验证。
有时需要对分析方法进行全面或部分的再验证。当原料药合成工艺发生改变时,可能引入新的杂质,杂质检查方法和含量测定方法的专属性就需要再进行验证,以证明有关物质检查方法能够检测新引入的杂质,且新引入的杂质对主成份的含量测定应无干扰。当分析方法发生部分改变时,如检测波长发生改变,则需要重新进行检测限、专属性、准确度、精密度、线性等内容的验证,以证明改变后的分析方法的合理性、可行性。


